CONGENITAL HYPERINSULINISM
Congenital hyperinsulinism of infancy is a disease of newborn children where life-threatening hypoglycemia (low blood glucose) arises because insulin secretion from the pancreas is inappropriately elevated. We are studying families with this condition. By genome-wide genetic screening in one such family, we discovered that mutations in the HADH gene can cause the disease. These mutations lead to deficiency of short-chain hydroxy-acyl-dehydrogenase (SCHAD), an enzyme which participates in mitochondrial fatty acid oxidation. Studies on genetic HADH variants and the disease mechanism are performed in cellular and mice models.
We have also characterized the mutation spectrum of the sulfonylurea receptor-1 gene (ABCC8), which is the most commonly mutated gene causing congenital hyperinsulinism in Norway. Diagnostic screening is offered for mutations in the ABCC8 gene as well as in other genetic causes of congenital hyperinsulinism (KCNJ11, GLUD1, GCK, HADH, HNF4A, HK1). We are currently performing a study of all congenital hyperinsulinism cases referred to Haukeland University Hospital during the last two decades.
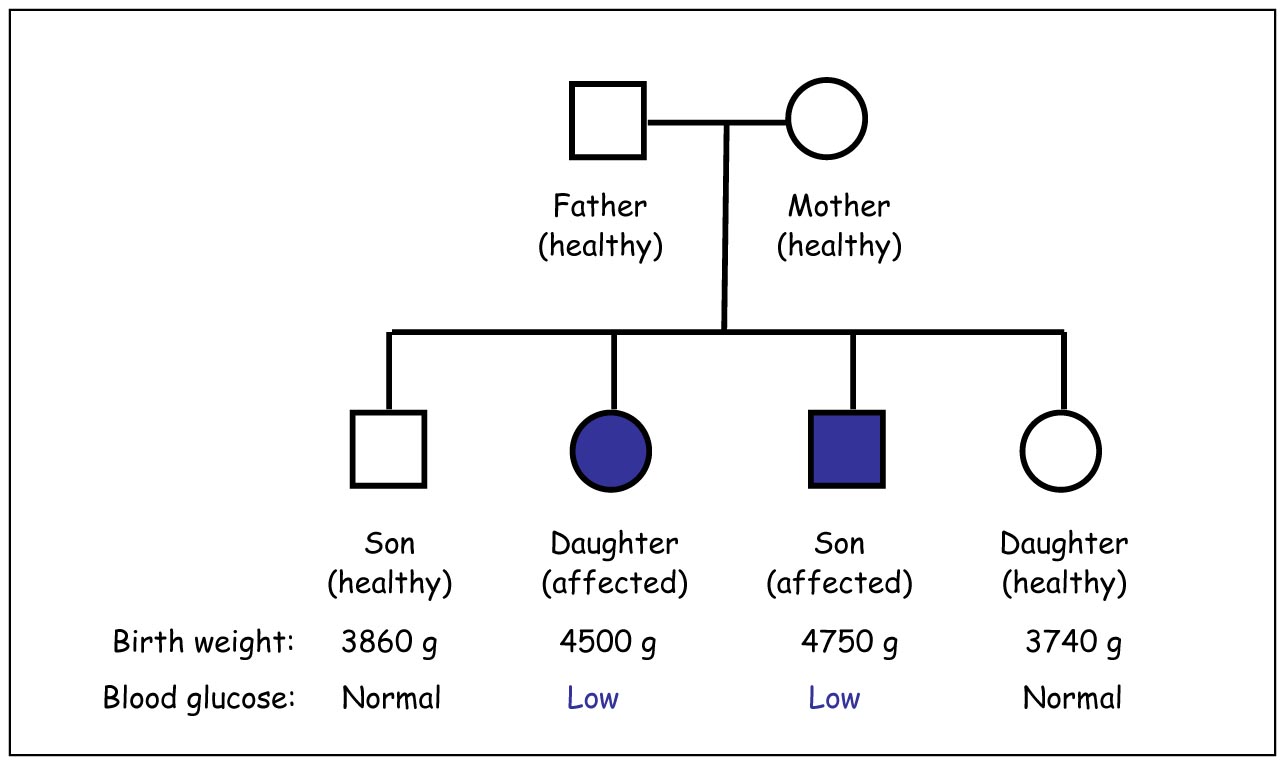
Example of a family with congenital hyperinsulinism of infancy. Family trees
like this are an important tool when determining which genes to examine.
Current participants / main collaborators
Medical student Christoffer Drabløs Velde, Gade Laboratory for Pathology, UoBergen
Molecular biologist Janniche Torsvik, Gade Laboratory for Pathology, UoBergen
Researcher Kelly Velasco, Department of Biomedicine, UoBergen
Ass. prof. Diana Toivola, Åbo Akademi University, Turku, Finland
Professor Rohit N. Kulkarni, Joslin Diabetes Center, Harvard Medical School, Boston, USA
Professor Pål R. Njølstad, Section for Pediatrics, Dept. of Clinical Science, UoBergen
Publications
St-Louis JL, Velasco K, Slipp BA, Hu J, Helgeland G, Steine SJ, De Jesus D, Kulkarni RN & Molven A (2023). Deficiency of the metabolic enzyme SCHAD in pancreatic β-cells promotes amino acid-sensitive hypoglycaemia. Journal of Biological Chemistry 299: 104986.
Velasco K, St-Louis JL, Hovland HN, Thompson N, Ottesen Å, Choi MH, Pedersen L, Njølstad PR, Arnesen T, Fjeld K, Aukrust I, Myklebust LM & Molven A (2021). Functional evaluation of sixteen SCHAD missense variants: Only amino acid substitutions causing congenital hyperinsulinism of infancy lead to loss-of-function phenotypes in vitro. Journal of Inherited Metabolic Disease 44: 240-252.
Helgeland Ø, Vaudel M, Júlíusson PB, Holmen OL, Juodakis J, Bacelis J, Jacobsson B, Lindekleiv H, Hveem K, Lie RT, Knudsen GP, Stoltenberg C, Magnus P, Sagen JV, Molven A, Johansson S & Njølstad PR (2019). Genome-wide association study reveals dynamic role of genetic variation in infant and early childhood growth. Nature Communications 10: 4448.
Patti ME, Goldfine AB, Hu J, Hoem D, Molven A, Goldsmith J, Schwesinger WH, La Rosa S, Folli F & Kulkarni RN (2017). Heterogeneity of proliferative markers in pancreatic β-cells of patients with severe hypoglycemia following Roux-en-Y gastric bypass. Acta Diabetologica 54: 737-747.
Gjelberg HK, Hoem D, Verbeke CS, Eide J, Cooper JC & Molven A (2017). Hypoglycemia and decreased insulin requirement caused by malignant insulinoma in a type 1 diabetic patient: when the hoof beats are from a zebra, not a horse. Clinical Case Reports 5: 761-768.
Molven A, Hollister-Lock J, Hu J, Martinez R, Njølstad PR, Liew CW, Weir G & Kulkarni RN (2016). The hypoglycemic phenotype is islet cell-autonomous in short-chain hydroxyacyl-CoA dehydrogenase-deficient mice. Diabetes 65: 1672-1678.
Molven A, Helgeland G, Sandal T & Njølstad PR (2012). The molecular genetics and pathophysiology of congenital hyperinsulinism caused by short-chain 3-hydroxyacyl-CoA dehydrogenase (SCHAD) deficiency. Pp. 137-145 in Monogenic Hyperinsulinemic Hypoglycemia Disorders (Eds. Stanley CA, De Leon DD). Vol. 21 in Frontiers in Diabetes. Karger, Basel, Switzerland 2012. ISBN 978-3-8055-9943-6.
Pörksen S, Laborie LB, Nielsen L, Andersen ML, Sandal T, de Wet H, Schwarz E, Åman J, Swift P, Kocova M, Schönle EJ, de Beaufort C, Hougaard P, Ashcroft F, Molven A, Knip M, Mortensen HB, Hansen L, Njølstad PR & Hvidøre Study Group on Childhood Diabetes (2010). Disease progression and search for monogenic diabetes among children with new onset type 1 diabetes negative for ICA, GAD- and IA-2 antibodies. BMC Endocrine Disorders 10: 16 (1-10).
Sandal T, Laborie LB, Brusgaard K, Eide SÅ, Christesen HBT, Søvik O, Njølstad PR & Molven A (2009). The spectrum of ABCC8 mutations in Norwegian patients with congenital hyperinsulinism of infancy. Clinical Genetics 75: 440-448.
Christesen HB, Tribble ND, Molven A, Siddiqui J, Sandal T, Brusgaard K, Ellard S, Njølstad PR, Alm J, Jacobsen BB, Hussain K & Gloyn AL (2008). Activating glucokinase (GCK) mutations as a cause of medically responsive congenital hyperinsulinism: prevalence in children and characterisation of a novel GCK mutation. European Journal of Endocrinology 159: 27-34.
Molven A, Matre GE, Duran M, Wanders RJ, Rishaug U, Njølstad PR, Jellum E & Søvik O (2004). Familial hyperinsulinemic hypoglycemia caused by a defect in the SCHAD enzyme of mitochondrial fatty acid oxidation. Diabetes 53: 221-227.
Molven A, Rishaug U, Matre GE, Njølstad PR, Søvik O (2002). Hunting for a hypoglycemia gene: Severe neonatal hypoglycemia in a consanguineous family. American Journal of Medical Genetics 113: 40-46